Kutatók igazolták, hogy jelentősen eltért a variánsok diverzitása és a SARS-CoV-2 koronavírus terjedési mintázata az első két hazai járványhullámban. Amíg az első hullámban a korán meghozott járványügyi korlátozó intézkedések feltartóztatták a lakosság körében keringő változatok terjedését, addig a második hullám észrevétlenül indult: a járvány ezen szakaszáért döntően felelős vírusváltozat korán bekerült az országba, és hónapokon át észrevétlenül terjedt, mielőtt a járvány újból berobbant volna. Annak ellenére, hogy ez a vírusváltozat más változatokhoz képest nem volt fertőzőképesebb, véletlen események folytán mégis ki tudta szorítani azokat.
Lezárult az a járványügyi szempontból fontos hazai tudományos munka, amely közel másfél éve indult az ELKH Szegedi Biológiai Kutatóközpontban (SZBK) működő Biotechnológiai Nemzeti Laboratóriumban, a Magyar Molekuláris Medicina Kiválósági Központban (HCEMM), a Pécsi Tudományegyetem Virológiai Nemzeti Laboratóriumában, az Eötvös Loránd Tudományegyetem Genetikai Tanszékén és a Szegedi Tudományegyetem Bolyai Intézetének Egészségbiztonság Nemzeti Laboratóriumában. A Virus Evolution folyóiratban megjelent közlemény a koronavírus-járvány első két hullámának vírusgenetikai és terjedési jellegzetességeit tárta fel a betegektől származó, archivált vírusminták genomszekvenálása és a földrajzi elterjedést leíró elemzések segítségével.
Ahogy arról már a kutatás korábbi szakaszában írtunk (itt és itt), a magyarországi Covid-járványt meghatározó vírusváltozatok genetikai vizsgálata fontos eleme a nemzetközi vírusmonitorozásnak, ugyanis a feltárt mintázatok irányt mutatnak a jövőbeli járványügyi helyzetekre való felkészüléshez. Világviszonylatban a Covid-kutatások egyik kiemelt fókuszpontja a fertőzés terjedési láncolatának és a kórokozó természetszerű változásainak nyomon követése, tekintettel arra, hogy a vírus örökítőanyagának természetes változásai (mutációi) meghatározóak a patogén ágens életképessége és fertőzőképessége szempontjából.
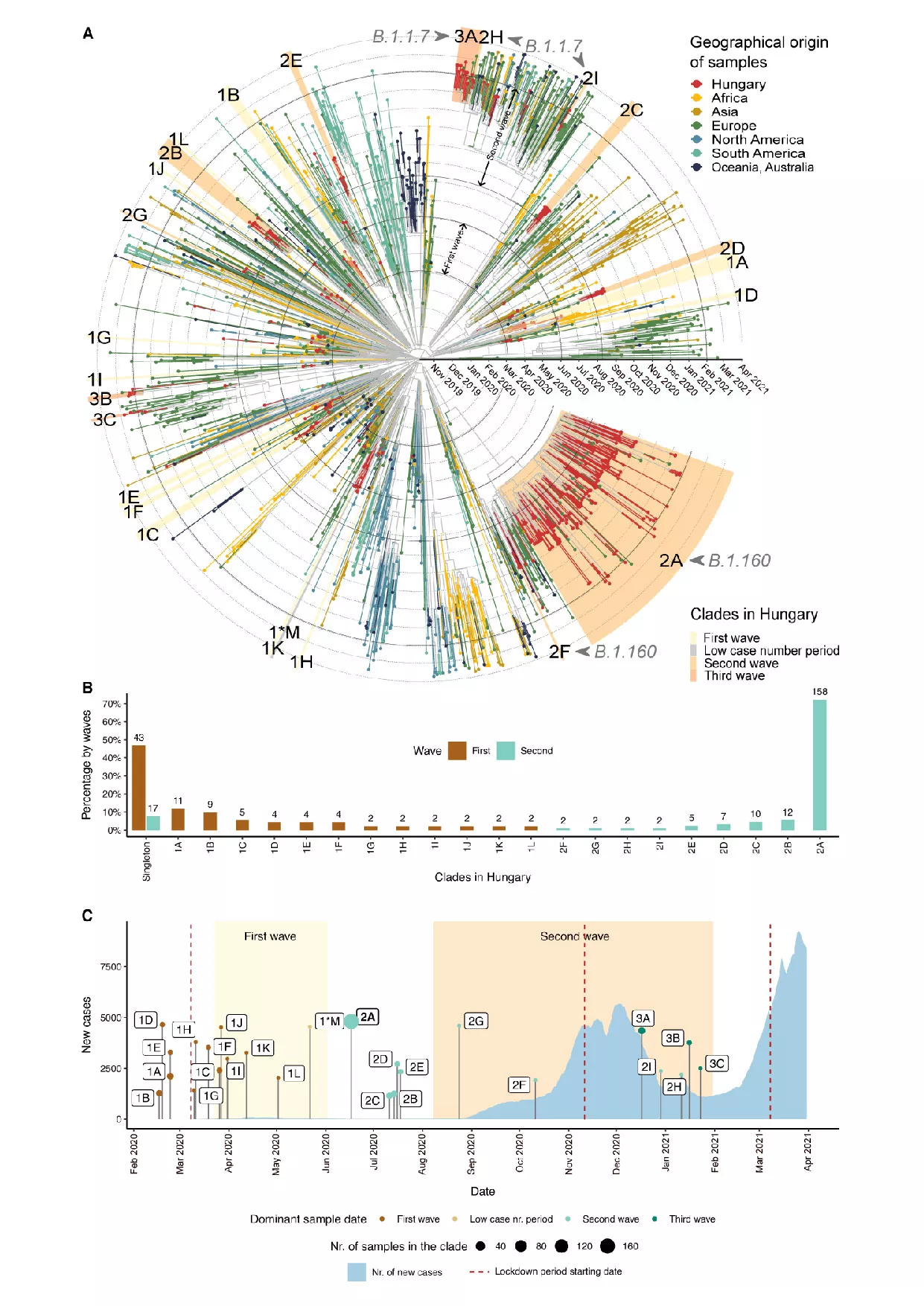
A kutatási projekt lezárulásáig a nemzetközi szinten is elismert kutatócsoport összesen 352 hazai minta genomszekvenciáját elemezte, a WHO által létrehozott és működtetett GISAID-adatbázis több mint 1300 nemzetközi mintájával összevetve (105 hazai minta a 2020 tavaszi első járványhullámból, 219 minta a 2020 őszén észlelt második hullámból származott). A két hullámban azonosított 25-féle vírusváltozat genetikai jellemzői alapján kirajzolódó úgynevezett evolúciós törzsfa elemzése azt igazolja, hogy a magyarországi első hullámot domináló változatok 2020 elején más európai országokból kerültek be hazánkba, és jellemzően lokális fertőzési gócokat alakítottak ki, mivel a gyors járványügyi intézkedések jelentősen korlátozták a vírus terjedését. Az elemzés arra is bizonyítékkal szolgál, hogy az első hullám észlelése előtt a koronavírus nem terjedt széleskörűen Magyarországon: az első hazai fertőzési láncok mindössze kb. két héttel korábban indulhattak, mint ahogy az első beteget azonosították. Ez markáns különbség számos nyugat-európai és észak-amerikai régióhoz képest, ahol hosszabb ideig „lappangva” terjedt a járvány.
Ezzel szemben a második hullám alattomosan indult: 2020 nyarán az összeurópai szinten feloldott határátlépési korlátozások nyomán újraindultak a nemzetközi utazások, és ennek következtében már röviddel a határnyitás után bekerült Magyarországra az a vírusvariáns, amely később országos szinten dominálta a járványt. Június elejétől szeptember közepéig azonban észrevétlenül terjedt ez a vírusvariáns a hazai lakosság körében – vélhetően olyan csoportokban, akiknél a kórlefolyás nem indokolt sem tesztelést, sem kórházi ellátást, így a „lappangva” terjedő esetek nem kerültek látótérbe. Az evolúciós törzsfa elemzése rámutat, hogy a hazai második hullámot domináló vírusvariáns (B.1.160) több nyugat- és közép-európai országban is meghatározó szerepet töltött be a koronavírus-járvány újabb fellángolásában. Általánosságban ez számított az egyik legelterjedtebb vírusváltozatnak Európában. A 2020 őszi-téli időszakban Magyarországon messze túlszárnyalta az összes egyéb vírusváltozat jelenlétét, és a világviszonylatban jellemző előfordulási gyakorisághoz képest 17-szer gyakrabban ez okozta a regisztrált megbetegedéseket. A kutatók azt is megvizsgálták, mi állhat a domináns vírusvariáns nagyfokú elterjedése hátterében. Az epidemiológiai modellezésből kiderült, hogy nem a B.1.160 variáns nagyobb fertőzőképessége, hanem véletlenszerű események és a külső körülmények együttállása magyarázza e változat dominanciáját a második hullámban. Ezzel szemben a harmadik hullámért felelős alfa variáns valóban a nagyobb fertőzőképessége okán tudott dominánssá válni és az akkor már szigorú járványügyi korlátozások ellenére is nagyszámú megbetegedést okozni, több ezer hazai halálos áldozattal.
Összességében a kutatás két, a hétköznapi intuícióval szembemenő mintázatot is feltárt. Először is kiderült, hogy a jóval kisebb méretű első hullám során több vírusváltozat keringett, mint a második hullámban. Másodszor, a nyár végén berobbant második hullámot nem a nyaralásból hazatérők által behurcolt vírusváltozatok indították el, hanem egy már korábban behurcolt és lappangva terjedő változat. Az eredmények rávilágítanak a modern járványügyi felügyeleti módszerek kiemelt szerepére, illetve hozzájárulnak a járványos megbetegedések lappangó terjedésének megértéséhez.