Combining the use of pharmacophores, the most general concept of representing drug-target interactions, with the theory of protein hotspots, Hungarian researchers led by the Medicinal Chemistry Research Group of the ELKH Research Centre for Natural Sciences have developed a novel design protocol for fragment libraries. The SpotXplorer approach compiles small fragment libraries that maximize the coverage of X-ray confirmed binding pharmacophores at the most preferred hotspots.
Fragment-based drug discovery (FBDD) has truly come of age, demonstrating its usefulness in many drug discovery programs, providing clinical candidates and multiple approved drugs. FBDD programs are routinely initiated by the screening of small and polar compounds (fragments) against the protein of interest. The reduced complexity of fragments provides more opportunity to make productive interactions with the target, however, their small size typically results in low affinity binding. Therefore, a library of fragments are screened in high concentration using mostly biophysical assays. Fragment screening libraries are generally compiled using ligand-based physicochemical criteria, such as molecular weight, lipophilicity, number of H-bond acceptors and donors, etc. In 2016, when the Innovative Training Network FRAGNET was launched with Iwan de Esch of VUA and Rod Hubbard of Vernalis and York University, many people considered the Rule of Three (molecular weight < 300, ClogP < 3, the number of hydrogen bond donors and acceptors each should be < 3 and the number of rotatable bonds should be < 3) as a general criteria for the design of fragment libraries.
At an annual FRAGNET meeting in Barcelona, researchers had a great discussion with Dan Erlanson, Chris Murray and Mike Hann, members of TTK’s advisory board, on the specificity and promiscuity of fragment binders. Recognising that some fragments appeared as hits more frequently than others, the team was prompted to analyse reported fragment hits in depth and identified a number of promiscuous fragments forming different interactions with the targets. Since their previous investigations on the thermodynamics of fragment binding revealed that fragments preferentially bind to protein hotspots (binding site regions with the largest contribution to the ligand binding free energy), they decided to combine the use of pharmacophores, the most general concept of representing drug-target interactions, with the theory of protein hotspots. Based on these concepts, they developed a new design protocol for fragment libraries.
In network technologies, a hotspot is a public wireless (WiFi) internet access point, through which anyone can connect to the Internet with a suitable device. Proteins found in the human body also have hotspots, which serve as access points for their endogenous ligands and interaction partners, and also for potential drugs. Knowing the location of the hotspot is important in our everyday communication, and even more so in drug discovery. Without knowledge on the hotspots of proteins, researchers can only find new drug molecules at random. As with the internet, however, the range of protein hotspots is limited, so accurate knowledge of their location is essential to identifying truly effective drugs. Using FTMAP, a validated hotspot mapping technology from the Vajda group at Boston University, researchers identified the hotspot of each protein with a co-crystallized fragment available in the PDB.
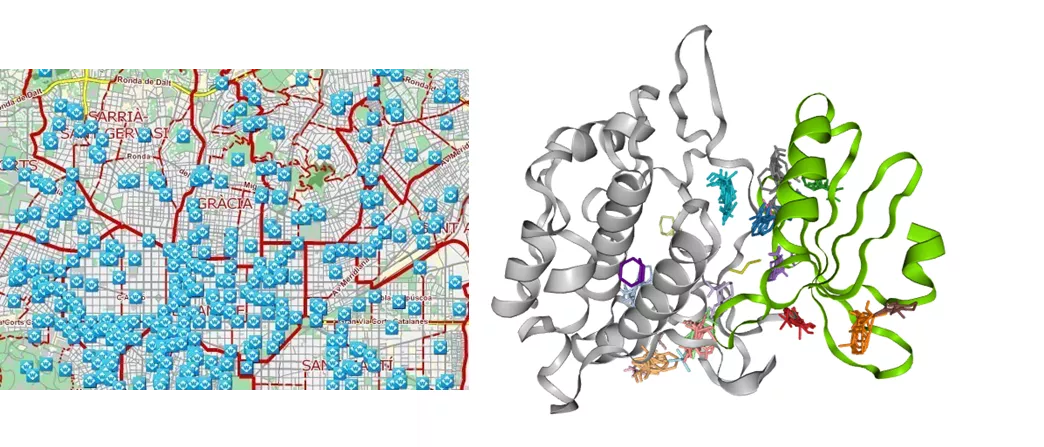
Hotspots available in central Barcelona and in the MELK kinase On the left: a higher number of blue rectangles indicates a better chance of connection. On the right: a higher number of coloured FTMap probes at a certain location indicates a stronger hotspot and better chance for fragments to bind. (PDB ID: 4IXP. Green and grey colours indicate the N- and C-terminus of the protein.)
The SpotXplorer technology is based on the recognition that hotspots of human proteins have been created by evolution to recognize molecules produced by our own body. Since humans only have a few thousand such molecules, researchers hypothesize that protein hotspots cannot show infinite variety. Just as one needs a user ID and password to connect to an internet hotspot, protein hotspots also identify related molecules. In the case of these molecules, the condition for binding is that the binding pattern of the molecule (spatial layout of H-bonding groups, aromatic rings, ionic centers, etc.) is recognized by the hotspot. Thus, by analysing the properties of hotspots known to date, binding patterns can be identified that will help find suitable starting points for fragments that effectively bind to disease-related target proteins.
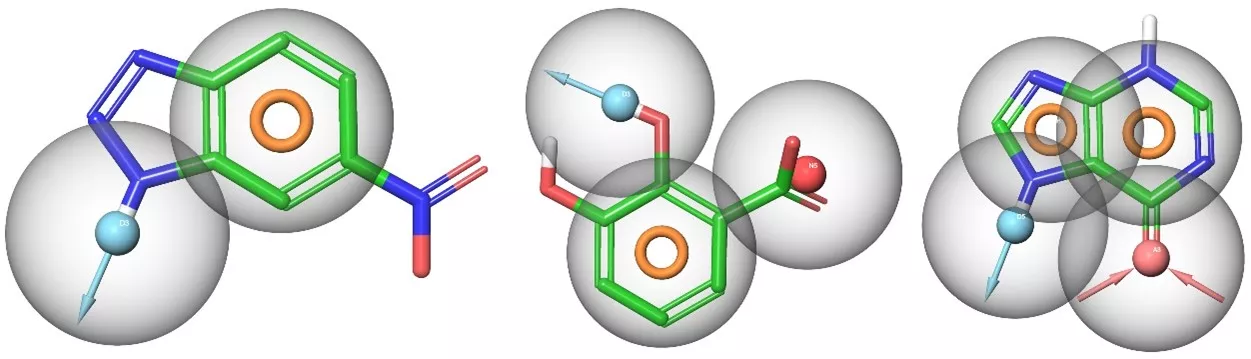
Typical 2-point, 3-point and 4-point pharmacophores identified at binding hotspots. Spheres indicate polar features (H-bond acceptors, donors and ionic centers), rings indicate aromatic features.
The researchers’ idea was to identify the set of binding patterns represented by fragment pharmacophores that covers the diversity of known hotspots and uses them to design a small, efficient fragment library to recognize this diversity. In contrast to previous, rule-based approaches, our design principles consider protein interactions to be responsible for fragment binding. Researchers therefore analysed critical interactions found at the hotspot of target proteins to derive fragment pharmacophores represented in fragment-protein complexes available in the PDB. Using this information, they designed a minimal diverse set of fragments (the SpotXplorer library) covering the majority of the experimental binding pharmacophores to be used for the identification of fragment starting points for drug discovery targets.

The SpotXplorer library contains 96 fragments representing 90% of the experimentally confirmed fragment binding pharmacophores. The library is available for screening at the Diamond Light Source, UK, and at RCNS, Hungary. (Illustration. Source: Centers for Disease Control and Prevention)
The designed collection of fragments available in the SpotXplorer library cover more than 90% of the currently known binding patterns. In this sense, SpotXplorer outperforms virtually all currently available fragment libraries, which are usually able to recognize less than half of the hotspot patterns. Therefore, it is likely that at least one of them is able to bind to the hotspot of almost any target protein. The SpotXplorer library was first validated against well-known drug targets represented by GPCRs and proteases, where the identified fragment hits retrieved an average of 70% of the known pharmacophores for those targets extracted from active molecules available in the ChEMBL database. Next, the team challenged the library against emerging drug targets, including the histone methyltransferase SETD2 involved in acute myeloid leukaemia and two SARS-CoV-2 proteins involved in the pathomechanism of COVID-19.

SpotXplorer fragment hit bound to the primary binding hotspot of the COVID-19 target NSP3 macrodomain. Binding pharmacophores, indicated here as blue spheres, are the key interacting features responsible for ligand binding.
SpotXplorer fragment hit bound to primary binding hotspot of the COVID-19 target NSP3 macrodomain. Binding pharmacophores, indicated here as blue spheres, are the key interacting features responsible for ligand binding.
Screening against SETD2 resulted in the first cell-permeable, non-nucleoside inhibitors that can be considered as viable starting points for further optimization. The XChem group at Diamond Light Source performed high throughput X-ray screening against the main protease (3CLPro) and the NSP3 macrodomain of SARS-CoV-2. These efforts yielded multiple fragment binders that were validated in both protein and cell-based assays, inhibiting the replication of the SARS-CoV-2 virus.
Overall, the researchers have developed a target-based library design protocol that provides fragment libraries for screening against relevant and emerging drug targets. Together with the viable chemical starting points, the technology provides the key pharmacophores for the target of interest encoded in each SpotXplorer fragment. Given the lack of structural data, this information would be especially useful for follow-up primary hits.
The paper is available at this link.
References:
Erlanson, Daniel A et al. “Twenty years on: the impact of fragments on drug discovery.” Nature reviews. Drug discovery vol. 15,9 (2016): 605-619. doi:10.1038/nrd.2016.109
Keserű, György M et al. “Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia.” Journal of medicinal chemistry vol. 59,18 (2016): 8189-206. doi:10.1021/acs.jmedchem.6b00197
Congreve, Miles et al. “A 'rule of three' for fragment-based lead discovery?.” Drug discovery today vol. 8,19 (2003): 876-7. doi:10.1016/s1359-6446(03)02831-9
Williams, Glyn et al. “Binding thermodynamics discriminates fragments from druglike compounds: a thermodynamic description of fragment-based drug discovery.” Drug discovery today vol. 22,4 (2017): 681-689. doi:10.1016/j.drudis.2016.11.019
Nature Comm paper https://doi.org/10.1038/s41467-021-23443-y (in press, currently under embargo)
Kozakov, Dima et al. “The FTMap family of web servers for determining and characterizing ligand-binding hotspots of proteins.” Nature protocols vol. 10,5 (2015): 733-55. doi:10.1038/nprot.2015.043